Thromboembolic Disease: Diagnosis and Treatment During Pregnancy
Authors
INTRODUCTION
Pregnancy and the puerperium present major challenges to women's hemostatic system. During implantation and early placentation, syncytiotrophoblasts penetrate the endothelium of maternal uterine capillaries, arterioles, and venules to establish the primordial or lacunar-type uteroplacental circulation. Subsequently, endovascular extravillous cytotrophoblasts invade uterine spiral arteries, orchestrating a remarkable morphological transformation to facilitate high-volume, low-resistance blood flow into the intervillous space. For successful pregnancy to occur, all of these events must occur in the absence of significant bleeding. Even more remarkable is the absence of potentially catastrophic maternal hemorrhage after separation of the placenta from the uterine wall following delivery. In this setting, 120–140 spiral arteries bereft of their muscle layer must thrombose during puerperal contractions. To meet these hemostatic challenges, pregnancy is accompanied by dramatic changes in both clotting and anticlotting factors that maximize hemostatic potential. However, a high price is paid to prevent pregnancy-associated hemorrhage because pregnancy and the puerperium are associated with a dramatically increased risk of venous thromboembolism (VTE).
Common manifestations of VTE in pregnancy include superficial and deep vein thrombosis (DVT), pulmonary embolus (PE), septic pelvic thrombophlebitis (SPT), and ovarian vein thrombosis (OVT). A leading cause of maternal morbidity and mortality, VTE occurs in from 1 in 1000 to 1 in 2000 pregnancies, representing a 10-fold increase in risk when compared to the risk in women of childbearing age.1, 2, 3, 4 Overall, more cases of thromboembolism probably occur in the antepartum period, however, given the shorter duration of the postpartum period the adjusted risk is 3–8 times higher in the postpartum period.5, 6
If a DVT remains untreated, PE will occur in 25% of the patients, with a mortality rate of 15%.7 Two thirds of these deaths occur within 30 minutes of the PE.8 In contrast, if treated promptly, less than 5% of patients with DVT will have a PE, and their mortality rate is less than 1%.9 These findings underscore the need to make a prompt diagnosis of DVT and expeditiously initiate therapy.
Numerous factors contribute to the development of VTE in pregnancy. Virchow's classic triad of stasis, hypercoagulability, and vascular trauma is present during pregnancy. Stasis of the lower extremities results from compression by the gravid uterus as well as the hormonally mediated increase in deep venous capacitance. Macklon and associates found progressive dilation of the deep veins of the leg during pregnancy with uterine compression of the inferior vena cava being greatest in the supine position and most decreased in the left lateral decubitus position.10 Estradiol may increase endothelium-derived nitric oxide, thus inducing dilation of capacitance vessels leading to venous stasis.11 Pregnancy is also a hypercoagulable state with an increase in several coagulation factors (I, II, VII, VIII, IX, X), a decrease in protein S,4, 6, 12 a progressive increase in resistance to activated protein C,13 and an increase in fibrinolytic inhibitors type-1 and type-2 plasminogen activator inhibitors (PAI-1 and PAI-2).14, 15 The risk of VTE is further increased in pregnant women with acquired or inherited thrombophilia.16, 17 Distention of the pelvic and lower extremity veins can lead to damage of the vascular endothelium and thrombosis. Operative vaginal delivery and particularly cesarean delivery clearly damage vessels.4 Cesarean delivery, in fact, is associated with a 9-fold increase in risk for VTE.5 This chapter defines the regulation of hemostasis, changes wrought by pregnancy, and the latest methods to diagnose, treat, and prevent VTE.
REGULATION OF HEMOSTASIS
Initiation of clotting
The primary initiator of hemostasis is tissue factor (TF), a cell membrane-bound glycoprotein.18, 19 Present on epithelial and stromal cells of diverse organs, TF is also present in high concentrations in amniotic fluid20 and uterine decidua.21, 22 While not normally expressed by endothelial cells19, 20 after vascular disruption, perivascular cell TF binds to plasma-derived factor VII.19 As demonstrated in Figure 1, the TF–factor VII complex can directly activate factor X or indirectly generate factor Xa by first activating factor IX, which can then complex with its cofactor, factor VIIIa, to activate factor X. Conversion of factor VII to VIIa by thrombin, or factors IXa, Xa, or XIIa, results in a 100-fold increase in activity.19 Once activated, factor Xa complexes with factor Va to convert prothrombin (factor II) to thrombin (IIa). Thrombin then cleaves fibrinogen to produce fibrin. Fibrin monomers self-polymerize and are covalently cross-linked by thrombin-activated factor XIIIa to form a stable hemostatic plug. Factor XI's role in hemostasis is to maintain, not initiate, clotting. Once TF–VIIa-induced clotting occurs, platelets aggregate in the fibrin clot. Factor XI binds to the surface of activated platelets, where it is activated by factor XIIa and, in turn, activates factor IX. Its adjuvant role in hemostasis is suggested by the minimal hemorrhagic sequelae attendant factor XI deficiencies,19 whereas excess factor XI results in 2.2-fold increased risk of thrombosis.23
| Fig. 1. TF initiates hemostasis by complexing with factor VII to directly convert factor X to Xa or indirectly generates Xa by converting factor IX to IXa, which, in turn, complexes with factor VIIIa to convert X to Xa. Factor Xa complexes with factor Va, to convert prothrombin (II) to thrombin (IIa). These processes are rapidly inhibited by the TF pathway inhibitor (TFPI). (Factors in italic denote negative effects.) However, factor XIa maintains clotting by serving as an alternative activator of factor IX on the surface of activated platelets. Thrombin cleaves fibrinogen to generate fibrin monomers that spontaneously polymerize and are covalently cross-linked by factor XIIIa to form a stable clot. Thus, true dampening of the clotting cascade requires inhibition of factor IXa- and Xa-mediated clotting. Activated protein C complexed with protein S (APC/S) serves this role by inactivating the factors VIIIa and Va. The most crucial endogenous anticoagulant system inactivates thrombin and Xa directly and includes α-2-macroglobulin, heparin cofactor II, and, most importantly, antithrombin (also called antithrombin III or ATIII). These antiproteases bind to heparin and vitronectin (Vn) as well as thrombin or Xa to shut-down the clotting cascade. Finally, fibrinolysis breaks down the fibrin clot. Fibrinolysis is mediated by tissue-type plasminogen activator (tPA), which binds to fibrin, where it converts plasminogen to plasmin. Plasmin, in turn, degrades fibrin but can be inactivated by α-2-antiplasmin embedded in the fibrin clot. Fibrinolysis is primarily inhibited by type-1 plasminogen activator inhibitor (PAI-1), the fast inactivator of tPA, which exists in its active form bound to Vn. The thrombin activatable antifibrinolytic system (TAFI) forms a second important antifibrinolytic system.
|
Endogenous anticoagulant system
As noted, thrombin is the ultimate arbiter of coagulation because it not only cleaves fibrinogen to fibrin but also amplifies the coagulation signal by activating factors V, VII, VIII, and XIII, as well as platelets.24 While critical to physiological hemostasis, inappropriate or excess thrombin generation can lead to fatal thrombotic diseases.25 To prevent such untoward effects, excess thrombin and factor Xa activity is rapidly inhibited by heparin cofactor II, α-2 macroglobulin, and antithrombin (AT), previously termed antithrombin III or ATIII (see Fig. 1).
Thrombin/factor Xa binds to each of these inhibitors and to the large plasma glycoprotein, vitronectin (Vn), to form a ternary (i.e., three-part) complex.26 A conformational change within the ternary complex facilitates binding to heparin, which augments the rate of thrombin inactivation by several thousand-fold.27, 28, 29 The most physiologically active of such complexes is thrombin–AT. Levels of thrombin–AT complexes increase throughout uncomplicated pregnancy indicating that pregnancy is associated with increased thrombin generation.30
Thrombin is also inactivated on the endothelial surface after its high-affinity binding to thrombomodulin. The resultant complex activates protein C which, when complexed with its cofactor, protein S, inactivates procoagulant factors VIIIa and Va to greatly limit thrombin generation31 (see Fig. 1). Levels of free protein S and protein S activity decrease approximately 40% during pregnancy, because of pregnancy-associated increases in the protein S carrier protein–complement 4b-binding protein.31, 32
A second level of regulation is via tissue factor pathway inhibitor (TFPI). This protein contains two functional domains that initially bind factor Xa and subsequently bind the TF–VIIa complex.33, 34 The resultant quaternary inhibitory complex greatly reduces the functional duration of the TF–VIIa complex, inhibiting subsequent thrombin generation (see Fig. 1).
Fibrinolytic system
Another level of inhibition of clot propagation is fibrinolysis. Tissue-type plasminogen activator (tPA) binds to the fibrin clot to generate plasmin. The latter cleaves fibrin to generate fibrin degradation products (FDPs) (see Fig. 1). Plasmin can be inactivated by α-2-antiplasmin embedded in the fibrin clot. However, the primary inhibitor of fibrinolysis is type-1 plasminogen activator inhibitor (PAI-1). This 50,000-molecular weight (MW) antiprotease acts as the fast inactivator of the plasminogen activators (see Fig. 1). The principal source of PAI-1 is the endothelium, where its synthesis can be stimulated by inflammatory cytokines.35 In pregnancy, the uterine decidua is an alternative rich source of PAI-1 synthesis36 while the placenta produces PAI-2.37
The PAI-1 molecule is maintained in its active form as a result of tight binding to vitronectin (Vn).38, 39 The PAI-1 molecule dissociates from Vn, followed by the appearance of inactive tPA–PAI-1 complexes.40, 41 Activated platelets also secrete PAI-1–Vn complexes, resulting in stabilization of active PAI-1 at sites of vascular injury and initial plug formation.41 Thus, Vn-stabilized PAI-1 derived from both endothelium and platelets adheres to the endothelial cell wall and fibrin clot to counteract tPA-mediated fibrinolysis and hemorrhage.41
Paradoxically, the PAI-1–Vn complex also inhibits thrombin, albeit at a slower and less efficient rate than AT42 (see Fig. 1). Thus, the PAI-1–Vn complex also prevents pathological extension of the clot.43 Heparin also markedly increases the efficiency of thrombin inactivation by PAI-1–Vn.26 Because thrombin and PAI-1–Vn are initially incorporated into the newly formed clot, the PAI-1–Vn complex both inhibits thrombus extension yet prevents premature fibrinolysis, thus right-sizing the fibrin plug.26, 44
A second key mediator of fibrinolysis is the thrombin-activatable fibrinolysis inhibitor (TAFI). When fibrin is degraded by plasmin, carboxy-terminal lysines are exposed. Plasminogen binds to these residues to promote fibrinolysis. TAFI cleaves these lysine residues to prevent fibrinolysis. As is the case for protein C, TAFI is activated by the thrombin–thrombomodulin (TM) complex.45 Moreover, as was the case for PAI-1, the thrombin–thrombomodulin complex can both inhibit clotting via the generation of activated protein C and inhibit fibrinolysis to also right-size the fibrin clot.
RISK FACTORS FOR THROMBOEMBOLISM IN PREGNANCY
Pregnancy-associated changes in hemostatic and fibrinolytic proteins
As noted, pregnancy is a hypercoagulable state. Fibrinogen and factors II, VII, X, VIII, and XII increase by 20–200%, while factors V and IX remain unchanged and factor XI levels decrease by 30%.31 In contrast, endogenous anticoagulant levels increase only minimally (TFPI, α-2-macroglobulin), remain constant (AT, heparin cofactor II, and protein C), or significantly decrease (protein S) in pregnancy.31 Concentrations of PAI-1 increase two- to three-fold in pregnancy.38 The net effect of these changes in the hemostatic system is to increase the tendency toward thrombus formation, extension, and stability. Normalization of the coagulation parameters occurs 3 weeks' postpartum.46
Clinical risk factors for VTE in pregnancy
Clinical risk factors for VTE include those unrelated to pregnancy such as trauma, infection, obesity, nephrotic syndrome, age older than 35, bedrest, orthopedic surgery, and a previous history of DVT or PE.16, 47 Smoking (odds ratio [OR]: 2.4) and previous superficial vein thrombosis (OR: 9.4) are also noted to be significant independent risk factors for DVT or PE during pregnancy and the postpartum period.48 Pregnancy-specific risk factors include cesarean delivery, increased parity, and postpartum endomyometritis.16 The former is the most serious risk factor; cesarean delivery is associated with a nine-fold increase in the risk of VTE compared with a vaginal delivery.5, 48
Acquired and inherited thrombophilia
Pregnant patients with acquired or inherited thrombophilia are at particularly high risk for VTE. The former include antiphospholipid antibodies (APA) while the latter include a wide variety of relatively common genetic conditions that predispose to thrombosis (Table 1).
Table 1. Risk of thromboembolism in pregnant patients with acquired and inherited thrombophilia
| Condition | Risk of Inheritance | Thrombosis |
| Antiphospholipid antibodies | None | Approx. 14% |
| Factor V Leiden | AD | 0.2% |
| Prothrombin mutation | AD | 0.5% |
| Antithrombin deficiency | AD | 50% |
| Protein C deficiency | AD | Approx. 4% |
| Protein S deficiency | AD | Approx. 4% |
| Elevated factors VII, VIII, and XI | AD | Approx. 1% each |
| Thrombocythemia | None | Moderate |
| Hyperhomocysteinemia | AD | 0.2% |
Risks extrapolated from general population and affected case frequencies assuming a risk of thromboembolism in pregnancy of 0.67/1000 (taken from references 49 and 50.)
ACQUIRED THROMBOPHILIA
APA account for approximately 14% of VTE events in pregnancy.16, 17 These antibodies also cause thrombocytopenia and various adverse obstetrical outcomes including stillbirth, fetal growth restriction, and severe preeclampsia. At least two distinct classes of APA are closely linked to VTE. The first was described by Conley and Hartman in 1952 who reported that two patients with lupus had biological false-positive serological tests for syphilis and a plasma inhibitor of in vitro phospholipid-dependent clotting assays.51 These APA were termed lupus anticoagulants (LACs), although it was subsequently discovered that most affected patients did not have lupus and that their principal clinical consequence was thrombosis not hemorrhage.52 The second class of prothrombotic APAs was discovered by Harris and associates53 and termed anticardiolipin antibodies (ACA).
Neither LAC nor ACA are true antiphospholipid antibodies because their epitopes are proteins complexed with anionic phospholipids, rather than an isolated phospholipid moiety. In the case of pure LACs not associated with ACA reactivity, the target protein is prothrombin-embedded in ethanolamine-enriched hexagonal phospholipid formations. The latter's unusual biologic properties alter prothrombin's tertiary structure rendering it immunogenic.
The principal epitope of ACA is the 57-kD anionic phospholipid-binding glycoprotein, β-2-glycoprotein-I (β2GPI).54 Because β2GPI also binds to prothrombin, 33% of ACAs also have LAC activity, while 67% of these anti-β2GPI-anionic phospholipid complex antibodies do not prolong in vitro clotting assays. Thus, not all LACs have ACA reactivity and not all ACA have LAC properties. ACA may also target other anionic phospholipid binding proteins including thrombomodulin, protein C, and annexin V.55
While APAs may arise from conditions associated with VTE and poor pregnancy outcomes (e.g., SLE), these antibodies directly promote placental and vascular thrombosis by interference with a variety of anionic phospholipid-associated anticoagulant proteins (e.g., β2GPI, annexin V, antithrombin, thrombomodulin, and proteins C and S).56, 57, 58 Because of the frequent side effects of the various treatments, meticulous diagnostic criteria are required to diagnose both LAC activity and ACA reactivity.59 It also appears that the type of antibody as well as the titer may predict pathogenicity. In one study, patients with IgM ACA antibodies and low-positive IgG ACA antibodies had medical complication rates no different than patients with a negative antibody screen, whereas patients with LAC or IgG ACAs more than 19 GPL units had a four-fold higher rate of medical complications.57
HERITABLE THROMBOPHILIAS
Activated protein C resistance (APCR) occurs in 5% of the general population but in up to 40% of patients with VTE.58, 59, 60 The condition arises from a point mutation in factor V at its site of cleavage (inactivation) by protein C. Specifically, there is a substitution of a glutamine for an arginine at position 506 in the factor V polypeptide. This condition is primarily inherited in an autosomal dominant fashion and is designated the factor V Leiden (FVL) mutation.61, 62, 63 Compared with a general pregnant population with a thrombotic risk of 1 in 1400, the risk of thrombosis for pregnant women with FVL is 1 in 400–500.12 Pregnancy-associated reductions in protein S levels and increases in factor VIII exacerbate the prothrombotic effect of FVL. Patients with a single mutation have a 5–10-fold increased risk of VTE, whereas the rare homozygote-deficient patient has more than a 100-fold risk.64, 65, 66 However, while FVL is present in 40% of pregnant patients with VTE, given the low incidence of thrombosis in pregnancy (0.67/1000) and the high prevalence of the mutation in European populations (5%), the estimated risk of VTE among pregnant patients heterozygous for the mutation is only 0.2%.67 Tests are now readily available to screen for APCR. However, the presence of APAs, low protein S levels (as occur in pregnancy), and elevated factor VIII levels may cause false-positive laboratory results.16 Therefore, abnormal results should be confirmed by detection of the FVL mutation by polymerase chain reaction.
A mutation in the promoter of the prothrombin gene (G20210A) leads to overexpression of prothrombin with increased circulating levels and an increased risk of VTE.67 Present in 2–3% of the European population, it accounts for 17% of VTE events in pregnancy.67 However, again because of the low incidence of VTE in pregnancy, the actual risk of clotting in a carrier of this mutation is only 1 in 200 or 0.5%.67 Patients who are homozygous for this mutation as well as FVL homozygous are at very high risk for VTE in pregnancy (9%). In contrast, patients who are compound heterozygous for the FVL and prothrombin gene mutations have a lower, 4.6%, risk for VTE.49
The other inherited thrombophilias include autosomal-dominant (AD) deficiencies of AT, protein C, and protein S, and elevated levels of factors VII, VIII, and, as noted, XI. By far, the most serious is AT deficiency that is associated with antepartum and postpartum VTE in 50% of affected patients49 and despite its rarity (1 in 1000) may account for up to 20% of such cases in pregnancy.67 Combined, protein C and protein S deficiencies, although present in only 0.2–0.5% of the general population, account for 10–25% of VTE in pregnancy.67, 68 There is an increased risk of pulmonary infarctions associated with sickle cell hemoglobin C in pregnancy.69 Other conditions associated with an increased risk of VTE include elevated factor VII, VIII, and XI levels, as well as thrombocythemia and hyperhomocystinemia. Each confer modestly increased risks (two- to three-fold) but may, in aggregate, account for 10–20% of cases.24, 53, 67 It is unclear to what extent homozygosity for the newly described PAI-1 4G/4G allele, which causes increased PAI-1 levels, poses a risk for VTE.
Laboratory workup to exclude acquired and heritable thrombophilia
It may be reasonable to evaluate patients with newly diagnosed VTE or a personal or strong family history of VTE for acquired and heritable thrombophilia. Studies should rule out FVL mutation, prothrombin mutation 20210A, AT deficiency, protein C and protein S deficiency, LAC, ACA, anti-β2GPI antibodies, hyperhomocystinemia (homozygosity for the MTHFR mutation), and thrombocythemia.
DIAGNOSIS AND TREATMENT OF DEEP VEIN THROMBOSIS AND PULMONARY EMBOLISM
Deep vein thrombosis
The diagnosis of deep vein thrombosis (DVT) has typically been made on the clinical grounds of a tender and acutely swollen lower extremity in the absence of trauma. Homan's sign, which is pain in the calf when the foot is passively dorsiflexed, may be demonstrated. Unfortunately, many of these same symptoms are present in women with a normal pregnancy. More than 50% of patients exhibiting the classic presentation of calf or thigh tenderness, pain, erythema, and edema do not have a DVT.68, 70 An objective evaluation is required in all clinically suspicious cases.
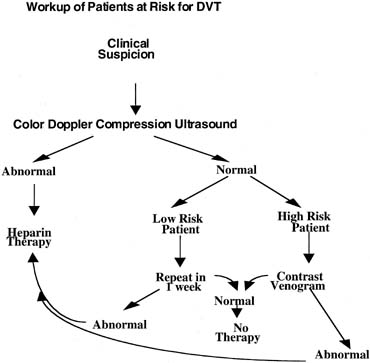
Although venography is considered the definitive test, noninvasive testing has emerged as the initial step in the diagnostic management of these patients (Fig. 2).
The first step when the diagnosis of DVT is being considered may be a D-dimer assay. The negative predictive value of this test may effectively reduce the clinical suspicion of DVT. D-dimer is the byproduct of degradation of fibrin by plasmin and these products may be tested using monoclonal antibodies specific to the D-dimer molecule; levels will be elevated when the coagulation system is activated. Thus, the test is limited by factors that may increase D-dimers, including pregnancy, the postpartum and postoperative periods, and superficial thrombophlebitis. Studies in nonpregnant patients have supported its use as a "rule-out" test because of a sensitivity and negative predictive value.71, 72, 73 D-dimer testing in pregnancy may have an even stronger value as a "rule-out" test given the higher rate of false-positives with Homan's sign in pregnancy. Thus, in a pregnant patient with suspected DVT, the D-dimer as a first step may put the patient into a low risk category and reduce the need for further testing. A positive D-dimer assay in a pregnant patient, however, will require further evaluation.
|
ULTRASOUND
The most common modality used to evaluate DVT is lower extremity venous ultrasonography. Color Doppler may be used to evaluate vessel anatomy, blood flow, augmentation of blood flow with muscle activity, and vessel compressibility. Ultrasound is both highly sensitive (92%) and specific (98%) in popliteal and femoral vein thrombosis. Doppler flow studies are less effective for evaluating calf vein thrombosis with a sensitivity of only 50%.74, 75 If the patient is placed in the left lateral decubitus position, Doppler flow variation with respiration can assist in diagnosing isolated iliac vein thrombosis.10 Noncompressibility of the venous lumen in a tranverse plane under gentle probe pressure using duplex and color flow Doppler imaging is the most accurate critierion in the diagnosis of DVT.76 If the sonographic findings are abnormal, venous thrombosis can be diagnosed and treatment initiated. If the sonographic findings are normal and the patient has no other risk factors (e.g., no history of VTE, inherited thrombophilia, plasma D-dimers, or clinical progression), the study can be repeated in 7 days.77 If the sonographic findings are normal but there is a high index of suspicion (positive personal or family history, D-dimers, and clinical progression), a contrast venography procedure should be performed.
IMPEDANCE PLETHYSMOGRAPHY
Rarely used today, impedance plethysmography (IPG) was the first noninvasive test for DVT in pregnancy. It measures the electrical impedance between two electrodes wrapped around the calf. A pneumatic cuff applied to the upper leg is used to reduce venous outflow. Characteristic changes in impedance are noted on deflation of the occlusive cuff if no obstruction to outflow is present.78 IPG is reliable for evaluating for thrombus formation from the iliac vein to the knee. Compression by the enlarging gravid uterus may produce false-positives.79 Placing the patient in the left lateral decubitus position increases specificity.80 IPG was shown to be inferior to ultrasound in a comparison study of 985 symptomatic patients.81
MAGNETIC RESONANCE IMAGING
With more experience, magnetic resonance imaging is becoming a more useful modality in the diagnosis of DVT, with a sensitivity and specificity of nearly 100% and an accuracy of 96%.82, 83, 84 It is most useful for detecting thigh and pelvic vein thrombosis.85 Although the safety of MRI in pregnant women has yet to be proven, no adverse effects have been noted and the test does not involve ionizing radiation like venography.80 As more experience with this diagnostic modality is gained, it may find increasing use in the obstetric population.
VENOGRAPHY
Contrast venography remains the gold standard for diagnosing DVT, though MRI may be of equal value. In venography, contrast agents are injected into lower extremity veins and the venous system of the leg and pelvis are evaluated radiographically. Venography with an abdominal lead shield exposes the fetus to very low levels of radiation (0.0005 Gy).86 This exposure is below that associated with childhood cancers and teratogenicity.87, 88 The incidence of chemical phlebitis is 3%.89
If D-dimer has not effectively ruled out DVT, Doppler ultrasound is recommended as the initial imaging test in pregnant women with suspected DVT. If this noninvasive test is positive for a DVT, then treatment should be started. With equivocal test results, contrast venography or MRI (for pelvic thromboses) should be performed (see Fig. 2).
Pulmonary embolism
Similar to the diagnosis of DVT, the clinical diagnosis of pulmonary embolism (PE) lacks sensitivity and specificity. It occurs in 1 in 2500 pregnancies and results in obstruction to pulmonary arterial blood flow, vasoconstriction of small arterial vessels, and a progressive loss of surfactant. The triad of dyspnea, pleuritic chest pain, and hemoptysis occurs in only 25% of patients with documented PE.90 Other clinical manifestations include cyanosis, tachypnea, syncope, diaphoresis, fever, a pleural friction rub, and a fixed S2. Although arterial blood gases may show hypoxia, one in six patients with a PE will have a normal PaO2.91 It also should be remembered that the supine position may lower PaO2 by as much as 15 mmHg in the third trimester of pregnancy.92 An electrocardiogram may reveal right bundle branch block, right axis shift, Q wave in leads III and aVF, S wave in leads I and aVL more than 1.5 mm, T wave inversions in leads III, and aVF, or new-onset atrial fibrillation.93 Echocardiographic findings may include right ventricular dilation and hypokinesis, tricuspid regurgitation, and pulmonary artery dilation.
As in the evaluation of DVT, D-dimer is a sensitive, but not specific, test for PE and can be useful in the initial assessment for PE. A negative D-dimer concentration (<500 ng/mL), measured by a sensitive ELISA test, is associated with a 95% negative predictive value for the diagnosis of PE in nonpregnant patients.82 This high sensitivity is supported by a meta-analysis reviewing the utility of D-dimer in the diagnosis of PE; in nonpregnant patients the test shows a sensitivity of 95% (though the specificity is 45%), with a strong negatively likelihood ratio of 0.11.94 Physiologic increases in D-dimer in pregnancy make its utility in pregnant patients for ruling out disease even more effective; when a D-dimer is normal, there is a very low probability that the patient has a PE.
Pulmonary angiography is considered the gold standard in the diagnosis of PE. When the diagnosis of PE is still in question after performing less invasive testing like ventilation/perfusion or CT scanning, this is the test of choice. However, angiography, which involves the intravenous injection of radio-opaque contrast dye through catheterization, is limited by contraindications (cardiopulmonary compromise, renal insufficiency, disseminated intravascular coagulation, thrombocytopenia, and coagulopathy) and risks (0.5% mortality risk, 3% serious complication rate).95
Before improvements in computed tomography (CT) scanning, ventilation/perfusion (V/Q) scanning was the primary initial approach to the investigation of patients with suspected PE.96 V/Q scanning is somewhat limited, however, as results must be interpreted in the context of clinical probability. Only normal (negative) and low-probability scans in the setting of a low clinical probability, and high probability scans in the setting of high clinical probability are considered diagnostic. Nondiagnostic scans (i.e., intermediate probability) or low-probability scans in high-risk patients (e.g., thrombophilics, positive D-dimer, ECG, and echocardiogram) should be followed-up with a color Doppler compression ultrasound study of the lower extremities. The presence of a DVT in conjunction with a nondiagnostic lung scan mandates therapy. However, fewer than 30% of unselected patients with PE have radiographic signs of DVT at the time of presentation.97, 98 Thus, a negative compression ultrasound in a high risk patient warrants pulmonary angiography77 (Fig. 3).
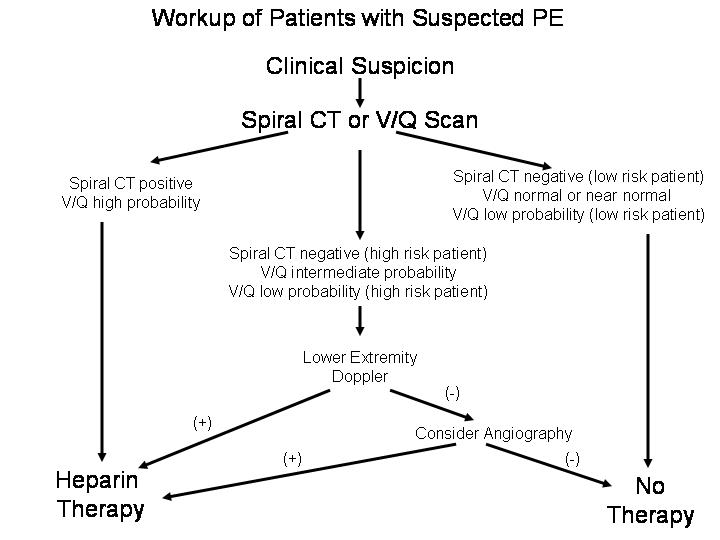 Fig 3: Workup of patients with suspected PE.
Fig 3: Workup of patients with suspected PE.
CT, specifically spiral CT, has evolved as a comparable, if not superior, modality to V/Q scanning in the evaluation of PE. This test involves the injection of intravenous radio-opaque contrast while simultaneously imaging the distribution of contrast in the pulmonary vasculature. This test is highly effective at diagnosing large segmental and central emboli, though it is more limited with subsegmental vessels. Unlike V/Q scanning, results require less interpretation within the context of pretest probability. A study in nonpregnant patients showed a high rate of definitive positive or negative diagnosis in spiral CT scanning (90% versus 54% for V/Q scanning), reducing the need for further testing, though the overall rate of PE diagnosis was the same.99 The reported sensitivities of spiral CT compared with the gold standard pulmonary angiography vary widely (64–93%), though a meta-analysis of over 23 studies demonstrated a low 3-month occurrence rate of subsequent VTE and fatal PE (1.4% and 0.51%, respectively) when CT was negative for PE.100 However, because of the risk of missing PE in the small distal or subsegmental vessels, a provider who has a high suspicion for PE in a patient with a negative spiral CT may consider following-up with a lower extremity ultrasound/Doppler or even pulmonary angiography to rule out a clot.
Radiation exposure is an important consideration in the workup of PE. Although the amount of radiation exposure from a V/Q scan is less than 0.005 Gy of radiation, in a 1998 survey, only 67% of respondents reported that they perform V/Q scans in pregnant patients.1 One may further reduce the radiation exposure by performing a perfusion scan first. A normal perfusion study requires no further testing. Spiral CT scans and pulmonary angiography, with appropriate abdominal shielding and using minimal fluoroscopy, expose the fetus to less than 50 millirads for the entire examination.
MRI is a potentially nonionizing radiographic modality for diagnosing PE. Specifically, magnetic resonance angiography (MRA) which uses an injection of gadolinium to define the pulmonary vasculature, has been assessed with mixed but promising results.101, 102 At this time, this technique should be reserved for highly specialized centers.
Septic pelvic thrombophlebitis
An uncommon complication of pelvic infection, septic pelvic thrombophlebitis (SPT) typically occurs in the postpartum; however, it has been reported to occur after gynecologic procedures. It is more common after cesarean than vaginal delivery. Thrombus formation in the pelvic veins is a result of pelvic infection. Multiple infected emboli may result from thrombolysis. Physical findings are nonspecific. The typical presentation is a patient who has spiking fevers that persist despite adequate antibiotic coverage. CT or MRI imaging may aid in the diagnosis. Although these techniques may be specific, a nonocclusive thrombus in the iliofemoral veins may not be detected.2 Some authorities3, 103 suggest that patients with a presumed diagnosis of SPT should start on a course of therapeutic anticoagulation. In many cases, the clinical response is considered therapeutic and diagnostic. Defervescence is expected in 48–72 hours. In the absence of a clinical response, the cause of the fever should be reassessed for other possible intra-abdominal complications such as an abscess or perforated viscus. The duration of treatment has been somewhat arbitrary. Some authors advocate therapy for 7–10 days, while others recommend a full 6 weeks of anticoagulation.5, 71 A recent study has called into question the role of anticoagulation in SPT.72 In a study of 14 patients with CT-documented SPT, eight received continued antibiotic therapy alone (ampicillin, gentamicin, clindamycin) and six received a combination of heparin and antibiotic therapy. The overall incidence of SPT in this population of 44,922 women was 1 in 3000 deliveries (1 in 800 for cesarean deliveries, 1 in 9000 for vaginal deliveries). The duration of fever was not different between the two groups.
Ovarian vein thrombosis
The incidence of ovarian vein thrombosis is 1 in 4000 deliveries.73 Some question whether ovarian vein thrombosis is a distinct entity from SPT. Several clinical manifestations distinguish the two processes. Ovarian vein thrombosis (OVT) typically presents with acute pain 2–3 days' postpartum (with or without fever). It occasionally can be mistaken for appendicitis in a postpartum patient. It also can occur in the absence of infection and has been described in antepartum patients.76 Thrombosis can be diagnosed with CT or MRI and is most commonly noted in the right ovarian vein.5 Treatment for OVT is the same as outlined for SPT.
Treatment of VTE in pregnancy
The treatment of VTE in pregnancy mirrors that of the nonpregnant patient with the exception of avoiding warfarin during the antepartum period. Anticoagulation with heparin is the therapy of choice for DVT with or without PE. Supplementary therapy with leg elevation and application of warm moist heat may decrease edema and provide some symptomatic relief. In the case of PE, careful attention should be placed on maintaining the maternal PaO2 more than 70 mmHg or O2 saturation of more than 95%.
Heparin
Heparin is a complex glycosaminoglycan that potentiates antithrombin activity, increases levels of factor Xa inhibitor,82 and inhibits platelet aggregation.83 Heparin does not cross the placenta, appears safe for the fetus, and does not enter breast milk.84, 94 Side effects of heparin therapy include hemorrhage, osteoporosis, and thrombocytopenia. Hemorrhage is more common with concomitant aspirin use, recent surgery, thrombocytopenia, and liver disease, and is less common when using low-molecular-weight heparin preparations.77 Heparin-induced thrombocytopenia occurs in 3% of patients and has two forms: the more common early-onset, transient, heparin-induced platelet aggregation for which therapy need not be interrupted and the rare IgG-mediated, heparin-induced thrombotic thrombocytopenia that occurs within 2 weeks of initiating therapy and mandates cessation of therapy.77 The occurrence of heparin-associated and induced thrombocytopenia appears far less common with low-molecular-weight heparin.99 In any case, platelet counts should be followed for the first 3 weeks of therapy. Heparin-induced osteoporosis is far more common when doses of more than 15,000 units per day are administered for more than 6 months.100
The goals of initial therapy are to maintain heparin levels of between 0.2 and 0.4 U/mL, or antifactor Xa activity between 0.6 and 1.0 U/mL or the aPTT between 1.5- and 2.5-times control. The dose required to achieve these goals will vary between individuals because of biological variability in heparin-binding proteins such as vitronectin, fibronectin, von Willebrand factor, platelet factor 4, and histidine-rich glycoprotein.77 Heparin should be administered with an intravenous loading dose of 150 U/kg for PE and 100 U/kg for DVT, followed by an infusion rate of 20 U/kg per hour. Doses are adjusted until the target aPTT, heparin, or antifactor Xa level is reached. The aPTT, heparin, or antifactor Xa level should be evaluated every 4 hours in the early stages of therapy. Note that the aPTT is not reliable in LAC patients and, therefore, heparin concentrations should be followed-up. Intravenous heparin should be administered for at least 5 days or until clinical improvement is noted.101 Thereafter, heparin may be administered subcutaneously 10,000–15,000 units every 8–12 hours, titrating to an aPTT of 1.5–2-times control (or heparin level of at least 0.2 U/mL) obtained 6 hours after the morning dose.84, 102
Alternatively therapeutic low-molecular-weight heparin may be used (e.g., dalteparin sodium [Fragmin, Upjohn] 200 antifactor Xa units/kg administered subcutaneously in divided doses twice per day − 5000 μg q.b.i.d. in a 50-kg woman). Enoxaparin (Lovenox, Aventis) 1 mg/kg twice per day may also be used. In the absence of data that monitoring therapeutic efficacy is not required during pregnancy, it is our practice to titrate the dose of low-molecular-weight heparin to a target therapeutic antifactor Xa levels of 0.6–1.0 U/mL assessed 4–6 hours after administration. Low-molecular-weight heparin appears safe and efficacious in pregnancy.95 However, given concerns with epidural hematoma formation, epidural and spinal anesthesia are contraindicated within 18 hours of low-molecular-weight heparin administration. For this reason, we recommend stopping therapy at 36 weeks, or earlier if preterm delivery is anticipated, and using unfractionated heparin until delivery. An advantage of using low-molecular-weight heparin is a lower risk of heparin induced thrombocytopenia, though platelets should still be followed in the first 2–3 weeks of use.
After 4 months of either therapeutic unfractionated or low-molecular-weight heparin therapy, the therapy may be switched to a prophylactic dose of unfractionated heparin (5000 units subcutaneously every 12 hours adjusted to produce an antifactor Xa level of 0.1–0.2 U/mL 6 hours after the injection), or low-molecular-weight heparin (e.g., dalteparin sodium [Fragmin] 2500 antifactor Xa units subcutaneously twice a day, or enoxaparin [Lovenox] 30 mg subcutaneously twice daily titrated to maintain antifactor Xa levels of 0.1–0.2 U/mL 4 hours after administration). Again, we recommend stopping therapy at 36 weeks and using unfractionated heparin to permit epidural and spinal anesthesia for delivery.
Management of the delivery period can be difficult in high-risk patients. Vaginal or cesarean delivery should not be accompanied by significant risks of treatment-related bleeding if the procedure occurs more than 4 hours after a prophylactic dose of unfractionated heparin. If the patient has been using therapeutic doses of unfractionated heparin an aPTT should be checked preoperatively. Protamine sulfate may be administered to reverse a markedly prolonged aPTT at the time of vaginal or cesarean delivery. Similarly, vaginal or cesarean delivery should not be accompanied by significant risks of treatment related bleeding if it occurs more than 12 hours after a prophylactic dose of low-molecular-weight heparin or more than 18–24 hours after a dose of therapeutic low-molecular-weight heparin. Antithrombin concentrates can be used in antithrombin deficient patients in the peripartum period. Heparin or low-molecular-weight heparin should be resumed 4–6 hours after a vaginal delivery and 8–12 hours after a cesarean delivery.
Oral anticoagulant therapy can be initiated postpartum by titrating the warfarin dose to maintain the patient's international normalized ratio (INR) at approximately 2–2.5. Note that warfarin has a more rapid inhibitory effect on levels protein C than on many of the clotting factors because of the former's shorter half-life (6 hours vs. 24–48 hours). Therefore, heparin should always be maintained during the initial 4 days of warfarin therapy and until a therapeutic INR is achieved to avoid warfarin-induced skin necrosis and paradoxical thromboembolism.
Therapeutic anticoagulation should be continued for at least 20 weeks from the time of VTE diagnosis. If the patient has delivered and this duration has been completed, prophylactic therapy should be continued during the postpartum period for 6–12 weeks after a DVT and 4–6 months after a PE or complex iliofemoral DVT.
Calcium supplementation (1500 mg orally every day) should be provided to all patients treated with heparin or low-molecular-weight heparin during pregnancy. In patients receiving more than 15,000 units of unfractionated heparin for more than 6 months, bone densitometry studies can be performed in the postpartum period. Evidence of significant osteoporosis should prompt referral to a reproductive or medical endocrinologist familiar with treating osteoporosis in premenopausal women. As noted previously, the benefit of anticoagulation in patients with SPT has been questioned.71
Warfarin
Warfarin gains its therapeutic anticoagulant effect from its ability to inhibit the action of vitamin K, which is a cofactor in the synthesis of the final molecular forms of factors VII, IX, X, and prothrombin.104 Warfarin, a small molecule loosely bound to albumin, crosses the placenta. A 33% risk of embryopathy is associated with exposure between 7 and 12 weeks' gestation. Stigmata include nasal hypoplasia, stippled epiphysis, and central nervous system abnormalities. The latter include dorsal midline dysplasia characterized by agenesis of the corpus callosum, Dandy–Walker malformation, midline cerebellar atrophy, and ventral midline dysplasia characterized by optic atrophy.95 Fetal and placental hemorrhage is also a major complication with warfarin use during pregnancy and especially with delivery.105 Vitamin K or fresh-frozen plasma can be used to reverse the effects of warfarin in the rare pregnant patient using this anticoagulant. The prothrombin time begins to normalize within 6 hours of a 5-mg dose of vitamin K (orally or subcutaneous). While larger doses may work more quickly, they will render the patient resistant to re-anticoagulation with warfarin.
One clinical situation that may warrant the use of warfarin in pregnancy is the rare patient with a mechanical heart valve in pregnancy. Because of the lack of adequate controlled trials, no consensus exists to guide the management of these patients. Older studies using heparin with predominantly older-generation prosthetic heart valves have shown an increase in thrombogenic complications including fatal maternal valve thrombosis.106, 107, 108, 109 No large clinical studies exist to guide in the use of low-molecular-weight heparin in pregnant patients with mechanical heart valves.107 However, the manufacturer of Lovenox (Aventis) specifically recommends against its use in this setting based on reports to the FDA of valvular thrombosis in pregnant women treated with Lovenox. The use of low-dose aspirin in adjunct to warfarin or heparin has been advocated based on a study of antithrombotic therapy in high-risk patients with mechanical valves.108 The risk of fatal maternal valve thrombosis may outweigh the risks of warfarin to the fetus between 12 and 36 weeks' gestation. A full discussion of these risks should precede conception in this select population. Women with mechanical heart valves who choose either unfractionated or low-molecular-weight heparin during the first trimester should realize that they will be at higher risk for thrombosis. These women should be maintained on an intravenous dose of unfractionated heparin sufficient to prolong the aPTT two- to three-times the control. After the first trimester and until near-term, the therapy for these women can be switched to warfarin. Because warfarin does not accumulate in breast milk and does not induce an anticoagulant effect, it is not contraindicated in breastfeeding mothers.
Thrombolytic therapy and surgery
Thrombolytic therapy with plasminogen activators (tPA, urokinase, and streptokinase) is relatively contraindicated in pregnancy because of the theoretical risk of massive abruption. No controlled studies exist examining the efficacy and safety of thrombolytic therapy in pregnancy. In a review of 172 pregnant patients treated with thrombolytic therapy, the maternal mortality rate was 1.2%, the fetal loss rate was 6%, and maternal complications from hemorrhage occurred in 8%.110 Currently, massive PE with hemodynamic instability should be the only indication for thrombolytic therapy in pregnant patients.111 Surgery, including embolectomy, should be reserved for life-threatening settings.
Inferior vena cava filters
Although rarely used in pregnant patients, inferior vena cava filters may be used in pregnant patients with the same indications as the nonpregnant population. These indications include patients with recurrent PE despite adequate anticoagulation, cases of pulmonary embolism, or ileofemoral DVT in a patient with a contraindication to anticoagulation such as recent surgery, hemorrhagic stroke, or active bleeding, the development of serious hemorrhagic complications with anticoagulant therapy.112, 113, 114, 115 Retrievable vena cava filters may prove ideal for pregnant patients requiring this therapy. Currently, they remain investigational.116
Prevention of venous thromboembolism
Prevention of VTE should be the goal of the obstetrician. It is important for the clinician to define the true risk of VTE in a particular pregnant patient. Among pregnant patients who have had a previous VTE during pregnancy, there is a 10% risk of recurrence.117 However, in general, among patients with a history of VTE before their current pregnancy, the risk of recurrence will vary with the presence of a thrombophilia or risk factors at the time of the previous VTE. Brill-Edwards and colleagues prospectively studied 125 pregnant women with a previous episode of VTE and withheld heparin until the postpartum period.118 They observed that three of the 125 women (2.4%) had a recurrent VTE event (95% confidence interval [CI]: 0.2–6.9%). However, there were no recurrences in the 44 women who had no evidence of thrombophilia and whose previous VTE was associated with a temporary risk factor (e.g., oral contraception, surgery, and pregnancy). In contrast, among the 51 women with a thrombophilia or unexplained previous VTE episode, or both; three (5.9%) had a recurrent VTE (95% CI: 1.2–16.2%). Other authors have also observed that thrombophilic patients with a previous VTE or in whom the VTE was unexplained (i.e., not associated with nonrecurring risk factors) have a substantially increased risk of recurrent VTE in pregnancy.68, 119 We recommend that patients having a previous DVT associated with a nonrecurring nonpregnant state (e.g., high-estrogen dose oral contraceptives or after orthopedic procedures or surgery) who are without other risk factors (e.g., identifiable thrombophilia, need for prolonged bedrest, obesity, superficial thrombophlebitis) do not appear to need prophylactic heparin therapy during pregnancy but should receive postpartum prophylaxis because 80% of pregnancy-associated fatal PEs occur in the postpartum period. In these patients, the individual risks and benefits must be weighed. Obviously thrombophilic patients or those with previous unexplained DVT or other major risk factors for VTE should have both antenatal and postpartum unfractionated or low-molecular-weight heparin prophylaxis. We would include a VTE occurring in a previous pregnancy as unexplained risk factor for VTE.
Historically, mini-dose heparin has been effective in preventing DVT in patients at risk. As noted, the standard dose of unfractionated heparin is 5000 units administered subcutaneously every 12 hours. However, some authors recommend following-up heparin levels to guide VTE prophylaxis during pregnancy.120 In a small study by Barbour and associates, heparin doses appropriate for prophylaxis varied from patient to patient and from one trimester to the next. They suggested following-up heparin levels in order to obtain adequate prophylaxis.121 Adequate heparin levels for prophylaxis have been defined as 0.1–0.2 U/mL. Studies also exist that document the safety and effectiveness of low-molecular-weight heparin for thromboprophylaxis. A dose of 40 mg subcutaneously per day of enoxaparin was adequate to prevent VTE in a prospective study of 69 pregnancies (61 women).121 Patients with antithrombin deficiency or who were homozygous for prothrombin or FVL mutations should receive therapeutic unfractionated heparin or low-molecular-weight heparin therapy during pregnancy and full anticoagulation in the puerperium regardless of their history of previous VTE.
Nonpharmacologic therapies aimed at preventing VTE in pregnancy include left-lateral decubitus positioning during the third trimester, graduated elastic compression stockings, and pneumatic compression stockings. The graduated elastic compression stockings have been shown to increase femoral vein flow velocity in late pregnancy;122 however, their role in decreasing VTE in pregnancy has yet to be defined. Pneumatic compression stockings improve blood flow and decrease stasis by using sequential cephalad compression. In a study by Nicolaides and associates, pneumatic compression of the lower extremities increased blood flow in the femoral vessels by 240%.123 By increasing plasminogen activator, pneumatic compression stockings also increase fibrinolysis.124 In a meta-analysis of moderate-risk surgery, they were shown to decrease the incidence of DVT by 60%.125 Because pneumatic compression stockings have no hemorrhagic risk associated with use and have been shown to be an effective means of DVT prophylaxis in gynecologic oncology surgery,126 it stands to reason that they would be an ideal device for prophylaxis in at-risk pregnant patients at prolonged bed rest or who are undergoing a cesarean delivery.
SUMMARY
Venous thromboembolic disease remains a leading cause of maternal morbidity and mortality. The clinical symptoms and signs of DVT and PE are often misleading and erroneous, and thus a systematic approach is necessary to make a timely and efficient diagnosis. D-dimer testing is helpful to help rule out disease; a negative D-dimer test in pregnancy effectively rules out the presence of VTE. DVT compression ultrasonography should be used with follow-up contrast venography if needed. In PE, the first step is usually V/Q scanning or spiral CT, depending on institutional protocols and preferences/experience. Occasionally, a pulmonary angiogram is required. Documented VTE in pregnancy requires therapeutic anticoagulation for at least 4–5 months or the duration of pregnancy, whichever comes first, with either unfractionated or low-molecular-weight heparin. Postpartum treatment with heparin or warfarin is required.
In women at increased risk for thromboembolism in pregnancy because of previous history of VTE, the presence of a thrombophilia or absence of an identifiable risk factor at the time of their initial thrombosis mandates antenatal prophylaxis with unfractionated or low-molecular-weight heparin followed by postpartum anticoagulation. Patients with antithrombin deficiency or those who are homozygous for the prothrombin or FVL mutations require therapeutic heparin or low-molecular-weight heparin therapy during pregnancy and full anticoagulation in the puerperium regardless of their history of previous VTE.
In contrast, patients with a previous VTE associated with a known nonrecurring risk factor (e.g., surgery, estrogen-containing contraceptive) who are without a maternal thrombophilia do not appear to require antenatal prophylaxis but should receive postpartum anticoagulation because this is the point of maximal risk for PE. However, we posit that if the patient's previous VTE occurred in a pregnancy, antenatal prophylaxis is required even in the absence of a known thrombophilia, as this should be considered a recurring risk factor.
All pregnant patients should attempt to avoid the supine position late in pregnancy. Although yet to be proven in clinical study, graduated elastic stockings may be a useful adjuvant in preventing thromboembolic disease in pregnant women. Consideration should be given to the use of pneumatic compression stockings in patients undergoing cesarean delivery who have lesser risk factors for VTE including obesity, prolonged bed rest, and anticipated prolonged surgery. These should be continued until the patient is ambulating.
REFERENCES
Treffers PE, Huidekoper BL, Weenink GH et al: Epidemiological observations of thrombo-embolic disease during pregnancy and inthe puerperium, in 56,022 women. Int J Gynaecol Obstet 21: 327–31, 1983 |
|
Stein PD, Hull RD, Kayali F et al: Venous thromboembolism in pregnancy: 21-year trends. Am J Med 117: 121–5, 2004 |
|
Jacobsen AF, Skjeldestad FE, Sandset PM: Incidence and risk patterns of venous thromboembolism in pregnancy and uerperium--a register-based case-control study. Am J Obstet Gynecol 198: 233.e1–7, 2008 |
|
Toglia MR, Weg JG: Venous thromboembolism during pregnancy. N Engl J Med 335: 108, 1996 |
|
Macklon NS, Greer IA: Venous thromboembolic disease in obstetrics and gynaecology: the Scottish experience. Scott Med J 41: 83–6, 1996 |
|
McColl MD, Ramsay JE et al: Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost 78: 1183, 1997 |
|
Wessler S: Medical management of venous thrombosis. Annu Rev Med 27: 313–319, 1976 |
|
Kakkar V: Prevention of venous thrombosis and pulmonary embolism. Am J Cardiol 65: 50c–54c, 1990 |
|
Villasanta U: Thromboembolic disease in pregnancy. Am J Obstet Gynecol 93: 142–160, 1965 |
|
Macklon NS, Greer IS, Bowman A: An ultrasound study of gestational and postural changes in the deep venous system of the leg in pregnancy. Br J Obstet Gynaecol 104: 191, 1997 |
|
Goodrich S, Wood JF: Peripheral venous distensibility and velocity of venous blood flow during pregnancy or during oral contraceptive therapy. Am J Obstet Gynecol 90: 740, 1964 |
|
Hellgren M, Blomback M: Studies on blood coagulation and fibrinolysis in pregnancy, during delivery and in the puerperium. Gynecol Obstet Invest 12: 141, 1981 |
|
Walker MC, Garner EJ et al: Changes in activated protein C resistance during normal pregnancy. Am J Obstet Gynecol 177: 162, 1997 |
|
Kruithof EK, Tran-Thang C et al: Fibrinolysis in pregnancy: A study of plasminogen activator inhibitors. Blood 69: 460, 1987 |
|
Gerbasi FR, Bottoms S et al: Increased intravascular coagulation associated with pregnancy. Obstet Gynecol 75: 385, 1990 |
|
Girling JC, de Swiet M: Thromboembolism in pregnancy; an overview. Curr Opin Obstet Gynecol 8: 458–463, 1996 |
|
Ginsberg JS, Wells PS et al: Antiphospholipid antibodies and venous thromboembolism. Blood 86: 3685–3691, 1995 |
|
Nemerson Y: Tissue factor and hemostasis. Blood 71: 1–8, 1988 |
|
Rapaport SJ: Regulation of the tissue factor pathway. Ann NY Acad Sci 614: 51–62, 1991 |
|
Lockwood CJ, Bach R, Guha A et al: Amniotic fluid contains tissue factor, a potent initiator of coagulation. Am J Obstet Gynecol 165: 1335–1341, 1991 |
|
Lockwood CJ, Nenerson Y, Guller S et al: Progestational regulation of human endometrial stromal cell tissue factor expression during decidulalization. J Endocrinol Metab 76: 231–236, 1993 |
|
Lockwood CJ, Nemerson Y, Krikun G et al: Steroid-modulated stromal cell tissue factor expression: A model for the regulation of endometrial hemostasis and menstruation. J Clin Endocrinol Metab 77: 1014–1019, 1993 |
|
Meijers JC, Tekelenburg WL, Bouma BN et al: High levels of coagulation factor XI as a risk factor for venous thrombosis. N Eng J Med 342: 696–701, 2000 |
|
Dennington PM, Berndt MC: The thrombin receptor. Clin Exp Pharmacol Physiol 21: 349–358, 1994 |
|
Seiler SM: Thrombin receptor antagonists. Semin Thromb Hemost 22: 223–232, 1996 |
|
Preissner KT, DeBoer H, Pannekoek H et al: Thrombin regulation by physiological inhibitors: the role of vitronectin. Semin Thromb Hemost 22: 165–172, 1996 |
|
Ill CR, Rouslahti E: Association of thrombin-antithrombin III complex with vitronectin in serum. J Biol Chem 260: 15610–15615, 1985 |
|
Preissner KT, Zwicker L, Muller-Berghaus G: Formation, characterization and detection of a ternary complex between S protein, thrombin and antithrombin III in serum. Biochem J 243: 105–111, 1987 |
|
deBoer HC, deGroot PG, Bouma BN et al: Ternary vitronectin-thrombin-antithrombin III complexes in human plasma: Detection and mode of association. J Biol Chem 257: 3243–3248, 1993 |
|
Delomore MA, Burrows RF, Ofosu FA et al: Thrombin regulation in mother and fetus during pregnancy. Semin Thromb Hemost 18: 81–90, 1992 |
|
Esmon CT: Molecular events that control the protein C anti-coagulant pathway. Thromb Haemostas 70: 29–35, 1993 |
|
Comp PC, Thurnau GR, Welsh J et al: Functional and immunologic protein S levels are decreased during pregnancy. Blood 68: 881–885, 1986 |
|
Girard TJ, Warren LA, Novotny WF et al: Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature 338: 518–520, 1989 |
|
Broze GJ, Warren LA, Novotny WF et al: The lipoprotein-associated coagulation inhibitor that inhibits factor VII-tissue factor complex also inhibits factor Xa: Insight into possible mechanism of action. Blood 72: 1467–1473, 1988 |
|
van Hinsbergh VW, Kooistra T, van den Berg EA et al: Tumor necrosis factor increases the production of plasminogen activator inhibitor in human endothelial cells in vitro and in rats in vivo. Blood 72: 1467–1473, 1988 |
|
Schatz F, Lockwood CJ: Progestin regulation of plasminogen activator inhibitor type-1 in primary cultures of endometrial stromal and decidual cells. J Clin Endocrinol Metab 77: 621–625, 1993 |
|
Estelles A, Gilabert J, Anar J et al: Changes in plasma levels of type 1 and type 2 plasminogen activator inhibitors in normal pregnancy and in patients with severe preeclampsia. Blood 74: 1332–1328, 1989 |
|
Van Meijer, Gebbink RK, Preissner KT et al: Determination of vitronectin binding site on plasminogen activator inhibitor 1 (PAI-1). FEBS Lett 352: 342–346, 1994 |
|
Seiffert D, Wagner NN, Loskutoff DJ: Serum-derived vitronectin influences the pericellular distribution of type 1 plasminogen activator inhibitor. J Cell Biol 111: 1283–1291, 1990 |
|
Preissner KT, Holzhuter S, Justus C et al: Identification and partial characterization of platelet vitronectin: evidence for complex formation with platelet-derived plasminogen activator inhibitor-1. Blood 74: 1989–1996, 1989 |
|
Salonen EM, Vaheri A, Pollanen J et al: Interaction of plasminogen activator inhibitor (PAI-1) with vitronectin. J Biol Chem 264: 6339–6343, 1989 |
|
Ehrlich HJ, Klein Gebbink R, Keijer J et al: Alteration of serpin specificity by a protein cofactor: Vitronectin endows plasminogen activator inhibitor 1 with thrombin inhibitory properties. J Biol Chem 265: 13029–13035, 1990 |
|
Stringer HAR, van Swieten P, Heijnen HFG et al: Plasminogen activator inhibitor-1 released from activated platelets plays a key role in thrombolytic resistance. Studies with thrombin generated in the Chandler loop Arterioscler Thromb 14: 1452–1458, 1994 |
|
Ehrlich HJ, Gebbink RK, Preissner KT et al: Thrombin neutralizes plasminogen activator inhibitor I (PAI-1) that is complexed with vitronectin in the endothelial cell matrix. J Cell Biol 115: 1773–1781, 1991 |
|
Booth NA: TAFI meets the sticky ends. Thromb Haemost 85: 1–2, 2001 |
|
Shaver DC: Thromboembolic disease. In Sciarra JJ (ed): Gynecology and Obstetrics, Vol 2, Chap 31. Philadelphia, Lippincott, 2004 |
|
Bonnar J: Venous thromboembolism and pregnancy. Clin Obstet Gynecol 8: 455–473, 1981 |
|
Danilenko-Dixon DR, Heit JA et al: Risk factors for deep vein thrombosis and pulmonary embolism during pregnancy and post partum: A population-based, case-control study. Am J Obstet Gynecol 184: 104–110, 2001 |
|
Conard J, Horellou MH, Van Dreden P. et al: Thrombosis and pregnancy in congenital deficiencies in AT III (sic), protein C or protein S: study of 78 women. Thromb Haemost 63: 319–320, 1990 |
|
Clagett GP, Anderson FA, Jr, Heit J et al: Prevention of venous thromboembolism. Chest 108 (4 Suppl): 312S-334S, 1995 |
|
Conley CL, Hartman RC: Haemorrhagic disorder caused by circulating anticoagulant in patients with disseminated lupus erythematosus. J Clin Invest 31: 621–622, 1952 |
|
Feinstein DI, Rapaport SI: Acquired inhibitors of blood coagulation. Prog Hemostasis Thromb 1: 75–95, 1972 |
|
Harris EN, Gharavi AE, Boey ML et al: Anticardiolipin antibodies: detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet 2: 1211–1214, 1983 |
|
McNeil HP, Simpson RJ, Chesterman CN et al: Antiphospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: beta-2 glycoprotein-1 (apolipoprotein H). Proc Natl Acad Sci 87: 4120–4124, 1990 |
|
Rand JH, Wu XX, Andree HAM et al: Pregnancy loss in the antiphospholipid-antibody syndrome-a possible thrombogenic mechanism. N Engl J Med 337: 154–160, 1997 |
|
Lockwood CJ, Rand JH: The immunobiology and obstetrical consequences of antiphospholipid antibodies. Obstet Gynecol Surv 49: 432–441, 1994 |
|
Silver RM, Porter TF et al: Anticardiolipin Antibodies: Clinical Consequences of Low Titers. Obstet Gynecol 87: 494–500, 1996 |
|
Koster T, Rosendaal FR, deRonde H et al: Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden thrombophilia study. Lancet 342: 1503–1506, 1993 |
|
Svensson PJ, Dahlback B: Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med 330: 517–522, 1994 |
|
deStefano V, Leone G: Resistance to activated protein C due to mutated factor V as a novel cause of inherited thrombophilia. Hematologia 80: 344–356, 1995 |
|
Bertina RM, Koeleman BPC, Koster T et al: Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 369: 64–67, 1994 |
|
Voorberg J, Roeise J, Koopman R et al: Association of idiopathic venous thromboembolism with single point mutation at Arg 505 of factor V. Lancet 343: 1535–1536, 1994 |
|
Zoller B, Dahlback B: Linkage between inherited resistance to activated protein C and factor V gene mutation in venous thrombosis. Lancet 343: 1536–1538, 1994 |
|
Vandenbrocke JP, Koster T, Breit E et al: Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 344: 1453–1457, 1994 |
|
Bloemenkamp KWM, Rosendaal FR, Helmerhorst FR et al: Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing third generation progestogen. Lancet 346: 1593–1596, 1995 |
|
Gerhardt A, Scharf RE, Beckman MW et al: Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med 342: 374–380, 2000 |
|
Kyrle PA, Minar E, Hirschl M et al: High plasma levels of factor VIII and the risk of recurrent thromboembolism. N Engl J Med 343: 457–462, 2000 |
|
Simpson FG, Robinson PJ, Bark M et al: Prospective study of thrombophlebitis and “pseudo thrombophlebitis. ” Lancet 1: 331–333, 1980 |
|
Cunningham FG, Pritchard JA, Mason R: Pregnancy and sickle cell hemoglobinopathies: results with and without prophylactic transfusions. Obstet Gynecol 62: 419–424, 1983 |
|
Ginsberg JS: Management of venous thromboembolism. N Engl J Med 335: 1816–1828, 1996 |
|
Bounameaux H, de Moerloose P, Perrier A et al: Plasma measurement of D-dimer as diagnostic aid in suspected venous thromboembolism: an overview. Thromb Haemost 71: 1–6, 1994 |
|
Heim SW, Schectman JM, Siadaty MS et al: D-dimer testing for deep venous thrombosis: a metaanalysis. Clin Chem 50: 1136–47, 2004 |
|
Wells PS, Anderson DR, Rodger M et al: Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med 349: 1227–35, 2003 |
|
Heijboer H, Buller HR, Lensing AWA et al: A comparison of real-time compression ultrasonography with impedance plethysmography for the diagnosis of deep vein thrombosis in symptomatic patients. N Engl J Med 329: 1365–1369, 1993 |
|
Wells PS, Hirsh J, Anderson DR et al: Accuracy of clinical assessment of deep vein thrombosis. Lancet 345: 1326–1330, 1995 |
|
Hirsh J, Hoak J: Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis (in consultation with the Council on Cardiovascular Radiology), American Heart Association. Circulation 93: 2212–45, 1996 |
|
Wells PS, Brill-Edwards P, Stevens P et al: A novel and rapid whole-blood assay for D-dimer in patients with clinically suspected deep vein thrombosis. Circulation 91: 2184–2187, 1995 |
|
Hull R, Raskob G, Carter C: Serial impedance plethysmography in pregnant patients with clinically suspected deep-vein thrombosis. Ann Intern Med 112: 663, 1990 |
|
Clarke-Pearson D, Jelovesek F: Alterations of occlusive cuff impedance plethysmography results in the obsteric patient. Surgery 89: 594, 1981 |
|
American College of Radiology: 1998 Standards, p457. 1998 |
|
Heijboer H, Buller H, Lensing AW et al: A comparison of realtime compression ultrasonography with impedance plethysmography for the diagnosis of deep vein thrombosis in symptomatic outpatients. N Engl J Med 329: 1365, 1993 |
|
Tapson VF, Carroll BA, Davidson BL et al: The diagnostic approach to acute venous thromboembolism. Clinical practice guideline. American Thoracic Society. Am J Respir Crit Care Med 160: 1043–66, 1999 |
|
Evans AJ, Sostman HD, Witty LA et al: Detection of deep venous thrombosis: prospective comparison of MR imaging andsonography. J Magn Reson Imaging 6: 44–51, 1996 |
|
Carpenter JP, Holland GA, Baum RA et al: Magnetic resonance venography for the detection of deep venous thrombosis:comparison with contrast venography and duplex Doppler ultrasonography. J Vasc Surg 18: 734–41, 1993 |
|
Spritzer C, Evans C, Kay H: Magnetic resonance imaging of deep vein thrombosis in pregnant women with lower extremity edema. Obstet Gynecol 85: 603, 1995 |
|
Ginsberg JS, Hirsh J, Rainbow AJ et al: Risk to the fetus of radiologic procedures used in the diagnosis of maternal thromboembolic disease. Thromb Haemost 61: 189–196, 1989 |
|
Brent RL: The effects of embryonic and fetal exposure to x-ray, microwaves, and ultrasound. Clin Obstet Gynecol 26: 484–510, 1983 |
|
Witlin AG, Sibai BM: When a pregnant patient has a thromboembolism. Contemp OB/GYN December, 8–98, 1996 |
|
Barbour IA: Current concepts of anticoagulation therapy in pregnancy. Obstet Gynecol Clin North Am 24: 499, 1997 |
|
Sasahara AA et al: The urokinase pulmonary embolism trial. Monograph no 39, p 60. New York, American Heart Association, 1973 |
|
McNeil BJ et al: Measures of clinical efficacy: III. The value of the lung scan in the evaluation of young patients with pleuritic chest pain. J Nucl Med 17: 163, 1976 |
|
Ang CK, Tan TH, Walters WA et al: Postural influence on maternal capillary oxygen and carbon dioxide tension. BJM 4: 201, 1969 |
|
Sreeram N, Cheriex EC, Smeets JLRM et al: Value of 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 73: 298–303, 1994 |
|
Stein PD, Hull RD, Patel KC et al: D-dimer for the exclusion of acute venous thrombosis and pulmonary embolism: a systematic review. Ann Intern Med 140: 589–602, 2004 |
|
Fedullo PF, Tapson VF: Clinical practice. The evaluation of suspected pulmonary embolism. N Engl J Med 349: 1247–56, 2003 |
|
Balan KK, Critchley M, Vedavathy KK et al: The value of ventilation-perfusion imaging in pregnancy. Br J Radiol 70: 338, 1997 |
|
Stein PD, Hull RD, Saltzman HA et al: Strategy for diagnosis of patients with suspected acute pulmonary embolism. Chest 103: 1553, 1993 |
|
Turkstra F, Kuijer PM, van Beek EJ et al: Diagnostic utility of ultrasonography of leg veins in patients suspected of having pulmonary embolism. Ann Intern Med 126: 775, 1997 |
|
Cross JJ, Kemp PM, Walsh CG et al: A randomized trial of spiral CT and ventilation perfusion scintigraphy for the diagnosis of pulmonary embolism. Clin Radiol 53: 177–82, 1998 |
|
Moores L, Jackson WJ, Shorr A, Jackson J. Meta-analysis: outcomes in patients with suspected pulmonary embolism managed with computed tomographic pulmonary angiography. Ann Intern Med 141: 866, 2004 |
|
Oudkerk M, van Beek EJ, Wielopolski P et al: Comparison of contrast-enhanced magnetic resonance angiography and conventional pulmonary angiography for the diagnosis of pulmonary embolism: a prospective study. Lancet 359: 1643–7, 2002 |
|
Moores L, Jackson WJ, Shorr A, Jackson J: Meta-analysis: outcomes in patients with suspected pulmonary embolism managed with computed tomographic pulmonary angiography. Ann Intern Med 141: 866, 2004 |
|
Meaney J, Weg J, Chenevert T et al: Diagnosis of pulmonary embolism with magnetic resonance angiography and conventional pulmonary angiography for the diagnosis of pulmonary embolism: a Prospective study. Lancet 359: 1643, 2002 |
|
Hirsh J: Oral anticoagulant drugs. N Engl J Med 324: 1865-–1875, 1991 |
|
Wessler S, Gitel SN: Warfarin. From bedside to bench N Eng; J Med 311: 645–652, 1984 |
|
Oakley CM: Pregnancy and prosthetic heart valves. Lancet 344: 1643, 1994 |
|
Gohlke-Barwolf C, Acar J, Oakely C et al: Guidelines for prevention of thromboembolic events in valvular heart disease. Study group of the working group on valvular heart disease of the European society of cardiology. Eur Heart J 16: 1320, 1995 |
|
Elkayam UR: Anticoagulation in pregnant women with prosthetic heart valves: A double jeopardy. J Am Coll Cardiol 27: 1704, 1996 |
|
Turpie AG, Gent M, Laupacis A et al: Comparison of aspirin with placebo in patients treated with warfarin after heart valve replacement. N Engl J Med 329: 524, 1993 |
|
Turrentine MA, Braems G, Ramirez MM: Use of thrombolytics for the treatment of thromboembolic disease in pregnancy. Obstet Gynecol Surv 50: 534, 1995 |
|
Ahearn GS, Hadjiliadis D, Govert JA et al: Massive pulmonary embolism during pregnancy successfully treated with recombinant tissue plasminogen activator: A case report and review of treatment options. Arch Int Med 162: 1221, 2002 |
|
Kempczinski RF: Surgical prophylaxis of pulmonary embolism. Chest 89 (5 Suppl): 384–385, 1986 |
|
Arbogast JD, Blessed WB, Lacoste H et al: Use of two Greenfield caval filters to prevent recurrent pulmonary embolism in a heparin-allergic gravida. Obstet Gynecol 84: 652–654, 1994 |
|
Hux CH, Wapner RJ, Chayen B et al: Use of Greenfield filter for thromboembolic disease in pregnancy. Am J Obstet Gynecol 155: 734–737, 1986 |
|
Narayan H, Cullimore J, Krarup K et al: Experience with Cardial inferior vena cava filter as prophylaxis against pulmonary embolism in pregnant women with extensive deep vein thrombosis. Br J Obstet Gynaecol 99: 637–640, 1992 |
|
Zwaan N, Lorch H, Kulke C et al: Clinical experience with tempory vena caval filters. J Vac Interv Radiol 9: 594, 1998 |
|
Tengborn L, Bergqvist D, Matzsch T et al: Recurrent thromboembolism in pregnancy and puerperium. Is there a need for thromboprophylaxis ?. Am J Obstet Gynecol 160: 90, 1989 |
|
Brill-Edwards P, Ginsberg JS et al: Safety of withholding heparin in pregnant women with a history of venous thromboembolism. N Engl J Med 343: 1439–1444, 2000 |
|
Granddone E, Margaglione M et al: Genetic susceptibility to pregnancy-related venous thromboembolism: Roles of factor V Leiden, prothrombin G20210A, and methylene tetrahydrofolate reductase mutations. Am J Obstet Gynecol 179: 1324–1328, 1998 |
|
Ginsberg JS, Greer I, Hirsh J: Use of antithrombotoc agents during pregnancy Chest 119 (1 Suppl): 122S, 2001 |
|
Barbour LA, Smith JM et al: Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 173: 1869–1873, 1995 |
|
Norgren L, Austrell C, Nillson L. The effect of graduated elastic compression stockings on femoral blood flow velocity during late pregnancy. Vasa 24: 282, 1995 |
|
Nicolaides AN, Fernandes E, Fernandes J et al: Intermittent sequential pneumatic compression of the legs in the prevention of venous stasis and postoperative deep vein thrombosis. Surgery 87: 69-76, 1980 |
|
Ljungner H, Bergqvist D, Nilsson IM. Effect of intermittent pneumatic and graduated static compression on factor VIII and the fibrinolytic system. Acta Chir Scand 147(8): 657-61, 1981 |
|
Wells PS, Lensing AW, Hirsh J. Graduated compression stockings in the prevention of postoperative venous thromboembolism. A meta-analysis. Arch Intern Med 154(1): 67-72, 1994 |
|
Einstein MH, Pritts EA, Hartenbach EM. Venous thromboembolism prevention in gynecologic cancer surgery: a systematic review. Gynecol Oncol 105(3): 813-9, 2007 |